
Pityriasis Lichenoides
Updated: Jul 12, 2010
Introduction
Background
Pityriasis lichenoides is a rare cutaneous disorder of unknown etiology. Pityriasis lichenoides encompasses a spectrum of clinical presentations ranging from acute papular lesions that rapidly evolve into pseudovesicles and central necrosis (pityriasis lichenoides et varioliformis acuta or PLEVA) to small, scaling, benign-appearing papules (pityriasis lichenoides chronica or PLC).1,2 Although historically, the term Mucha-Habermann disease has referred only to PLEVA, the term applies broadly to the entire spectrum of disease including PLC. A rare febrile ulceronecrotic variant has been reported, which is a severe form of PLEVA with high fever and marked constitutional symptoms. Lesions may self-involute and resolve completely over weeks, or new lesions occasionally may appear in crops, waxing and waning spontaneously for months to years thereafter.
Pathophysiology
Mucha-Habermann disease is not a vasculitic process despite reports of immunoglobulin and complement deposition in vessels. Fibrin is not present in the walls of vessels, and thrombi are not found in the lumen. A cell-mediated mechanism has been proposed based on a T-lymphocytic infiltrate with a cytotoxic/suppressor phenotype, diminished epidermal Langerhans cells, and a reduction of the CD4/CD8 ratio. CD30 (Ki-1) cells, which are associated with large cell lymphoma, have been identified in the infiltrate of both lymphomatoid papulosis and Mucha-Habermann disease, leading some authors to view this as a self-limited self-healing lymphoproliferative disease.3,4 One study suggests that pityriasis lichenoides is a form of a T-cell dyscrasia, based on the presence of intraepithelial atypical lymphocytes, phenotypic abnormalities, and TCR-gamma rearrangements.5
Frequency
United States
The incidence of Mucha-Habermann disease in the United States has not been reported.
International
In approximately 44,000 patients seen over 10 years in 3 catchment areas in Great Britain, 17 cases of PLEVA were diagnosed.
Mortality/Morbidity
A case series of 22 children revealed a mean duration in PLEVA of 1.6 months to complete resolution and a mean duration in PLC of 7.5 months. The natural tendency of the disease is to remit spontaneously, but some cases may wax and wane over years. Disease duration may be longer in adults. A rare severe variant of PLEVA presents with a sudden eruption of diffuse coalescent necrotic ulcerations associated with high fever.6 Patients may develop complications such as interstitial pneumonitis, abdominal pain, malabsorption, central nervous system involvement, bacteremia, sepsis, and rheumatic manifestations. T-cell receptor clonal rearrangements of lymphocytic infiltrates have been detected in patients with PLEVA. Occasional cases (<2%) have been reported to evolve into cutaneous lymphoma, although some reports may have represented misdiagnosis of lymphomatoid papulosis.7
Race
All races are affected. A racial predisposition has not been reported.
Sex
A male predominance has been reported in the pediatric population and in patients presenting with febrile ulceronecrotic Mucha-Habermann disease.
Age
Most patients present during the first 3 decades of life. Studies of children have shown a variable age of onset from 3-15 years, with a mean age of 9.3 years.
Clinical
History
The history is dependent on where an individual patient's manifestations fall on the spectrum of Mucha-Habermann disease. Note the following:
- Pityriasis lichenoides et varioliformis acuta: The common variant of PLEVA presents with the abrupt appearance of multiple papules on the trunk, buttocks, and proximal extremities. Papules rapidly progress to vesicles and hemorrhagic crusts. Minor constitutional symptoms may be present. A patient with febrile ulceronecrotic PLEVA presents with acute constitutional symptoms such as high fever, malaise, and myalgias. Lesions of PLEVA may be associated with burning and pruritus.
- Pityriasis lichenoides chronica: At the subacute end of the spectrum, PLC may develop over days. PLC also is distributed over the trunk, buttocks, and proximal extremities.
Physical
The clinical presentation of Mucha-Habermann disease spans a continuum that is a function of the acuity of onset. PLEVA and PLC are not distinct diseases, but rather, they are different manifestations of the same process, although the process is accelerated in PLEVA. Note the following:
- PLEVA presents acutely with 10-50 erythematous–to–reddish brown or purpuric round or ovoid lichenoid papules that are 5-15 mm in diameter. Many papules demonstrate a pseudovesicular summit evolving to a central necrosis and a hemorrhagic crust. Note the images below.
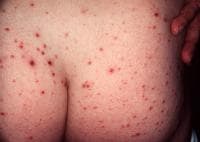
Typical hemorrhagic crusted papules of pityriasis lichenoides et varioliformis acuta.
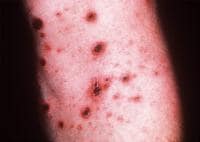
Close-up view of typical lesions of pityriasis lichenoides et varioliformis acuta.
- PLC presents as small erythematous–to–reddish brown papules, although with increased numbers compared to PLEVA. A fine scale usually is found, although not initially, which has been likened to frosted glass. The eruption often is polymorphic, with lesions at different stages of evolution. Note the images below.
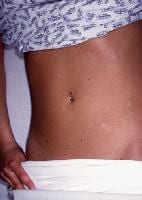
Scaling papules of pityriasis lichenoides chronica.
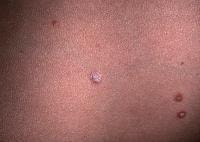
Close-up view of typical pityriasis lichenoides chronica lesions. Note papules in different stages of evolution and the scale with frosted-glass appearance in the lower right-hand corner.
- Lesions that are clinically consistent with both PLEVA and PLC often are found on physical examination, and the polymorphic appearance helps distinguish Mucha-Habermann from guttate psoriasis and lichen planus.
- PLEVA lesions may evolve into lesions of PLC.
- Most lesions heal with postinflammatory changes, such as a transient or persistent leukoderma or hyperpigmentation.
- Ulceronecrotic PLEVA presents with a sudden eruption of diffuse coalescent necrotic ulcerations associated with high fever. Some lesions may resemble those of PLC, but many are large, ulceronecrotic, and covered by a black oyster shell-like crust. Necrotic ulcerating lesions may eventuate into varioliform scars.
- Dark-skinned people rarely may present with widespread macular hypopigmentation rather than the typical papular morphology.8 This variant is most common in children. The diagnosis depends on careful inspection, which reveals subtle PLC lesions that are compatible histologically with this diagnosis.
- In both PLEVA and PLC, lesions are scattered but discrete.
- Lesions may be distributed symmetrically or asymmetrically on the trunk, buttocks, and proximal extremities, with occasional acral involvement. Lesions may appear on the palms, soles, face, and scalp. Asymmetric or segmental nondermatomal presentations have been reported.
- Erosions and hemorrhagic crusts may be found.
- Mucosal lesions consisting of irregular erythema and superficial ulcerations on the buccal mucosa and palate have been reported.
Causes
A number of acute exanthems (eg, Mucha-Habermann disease, pityriasis rosea, acute lichen planus, guttate psoriasis, erythema multiforme) are believed to be caused by a hypersensitivity reaction to infectious agents. Familial outbreaks, clustering of cases, and comorbid symptoms have been used to support these relationships in Mucha-Habermann disease, although clear causality is lacking. Elevations of pathogen-specific antibody titers also have been offered as proof of causality, but such immunologic responses may represent epiphenomena caused by nonspecific immune responses to unknown pathogens. The most commonly reported associated pathogens are Epstein-Barr virus (EBV), Toxoplasma gondii, and human immunodeficiency virus (HIV).
Two studies implicate EBV as an etiologic factor in Mucha-Habermann disease. The cases indicate that EBV may be a trigger in PLEVA, but neither study necessarily illustrates well-characterized comorbid EBV-mediated disease. Note the following:
Two studies implicate EBV as an etiologic factor in Mucha-Habermann disease. The cases indicate that EBV may be a trigger in PLEVA, but neither study necessarily illustrates well-characterized comorbid EBV-mediated disease. Note the following:
- In 1977, Boss et al reported a cluster of 10 cases seen over 1 year, in which eruptions were clinically consistent with PLEVA.9 Of these, 4 demonstrated elevated immunoglobulin G (IgG) complement-fixing antibodies to EBV. During resolution of the eruption, 3 of 4 patients demonstrated 4-fold or greater decrements in antibody titers.
- In 1989, Edwards et al described a child with a 3-week history of migratory arthralgias, monoarticular arthritis, acute pharyngitis, otitis media, and fevers to 104ºF.10 The girl developed a vesicular eruption localized primarily to the extremities, which clinically and histopathologically was consistent with Mucha-Habermann disease, with the exception of necrotic fibrin thrombi in the superficial and mid dermis. A Monospot test result was positive, and acute and convalescent serologies were consistent with a reactivation of EBV. Liver function tests were within normal limits. The patient's condition improved with treatment using oral tetracycline.
Elevated Toxoplasma gondii titers have been demonstrated in some patients with Mucha-Habermann disease. Despite an absence of clinical infection in case reports and series, more than 80% of primary Toxoplasmainfections are asymptomatic, and toxoplasmosis cannot necessarily be dismissed as a causative agent. Note the following:
- In 1969, Andreev et al were the first to suggest a link between toxoplasmosis and a recurrent PLEVA-like skin eruption in a patient with positive Toxoplasma serologies.11 Cutaneous lesions reportedly responded favorably to pyrimethamine.
- In 1972, Zlatkov and Andreev reported 11 patients with PLC and found that test results were positive for toxoplasmosis in 6 patients (55%) using complement-fixations test, intradermal test with toxoplasmin, and Sabin-Feldman dye test.12 Patients in whom test results were seropositive responded favorably to pyrimethamine, while no improvement was noted in the cutaneous lesions of 3 patients in whom results were seronegative.
- In 1987, Rongioletti et al described a patient who presented with acute onset of cutaneous lesions and histopathologic findings consistent with PLEVA.13 Serologic examination demonstrated enzyme-linked immunosorbent assay positivity for IgG and immunoglobulin M (IgM) and weak positive indirect fluorescence test results for IgM (1:16). Giemsa stain on the biopsy specimen failed to demonstrate Toxoplasma cysts. Spiramycin treatment was initiated, and lesions subsided over a few weeks. Convalescent serologies failed to demonstrate IgM 2 months later, although the authors still concluded that Toxoplasma species may have caused the cutaneous eruption.
- In 1997, Nassef and Hammam reported 22 patients diagnosed clinically and histopathologically with PLC and 20 healthy control subjects.14 Clinical examination for signs of toxoplasmosis only revealed axillary lymphadenopathy in 2 patients. Eight patients with PLC (36%) had a positive serodiagnosis by indirect hemagglutination versus 10% in the control group, and this difference was statistically significant. Using indirect immunofluorescence antibody tests, the difference was 36% versus 15%, respectively, but the difference was not statistically significant. All 22 patients with PLC were treated with pyrimethamine and trisulfapyrimidine, and lesions in 5 of 8 patients with seropositive results cleared completely within 2 months. None of the patients with seronegative results responded to treatment.
The first association between Mucha-Habermann disease and HIV infection was reported in 1991 by Ostlere et al.15 A patient with asymptomatic disease and a CD4+ T-cell count of 208 cells per microliter, diagnosed 6 months previously, presented with lesions consistent clinically and histopathologically with PLEVA. Note the following:
- In 1997, Smith et al reported a series of 5 patients with HIV infection in the early stage of disease, with CD4+ T-cell counts exceeding 200 cells per microliter and/or absolute lymphocyte counts within normal limits.16 The authors suggested that PLEVA serves as a marker of early–to–mid stage HIV disease.
- In 1998, Griffiths reported a patient who presented with a severely pruritic, erythematous, papular eruption that worsened as the CD4+ T-cell count fell from 200 to 20 cells/μ L.17 Biopsy confirmed PLC, and the disease progressed to febrile ulceronecrotic PLEVA. Dramatic improvement was attained using cyclosporine, and mild PLC-like lesions remained on maintenance doses. On saquinavir and lamivudine, the viral load became undetectable with a concomitant rise in the CD4+ count and a complete resolution of skin lesions. That the inherent immunologic dysregulation of HIV may play a role in Mucha-Habermann disease has been suggested.
In addition to EBV, Toxoplasma gondii, and HIV, a number of other infectious agents have been implicated. The following observations are provocative but may be chance associations. Note the following:
- Case reports have suggested that parvovirus B19 and adenovirus can trigger Mucha-Habermann disease.
- Herpes simplex has been associated with onset of the disease.
- One case report also describes resolution of PLC after tonsillectomy, with throat cultures yieldingStaphylococcus aureus and group A beta hemolytic streptococci.
- Piamphongsant similarly found coagulase-positive staphylococci on throat cultures in 4 of 10 patients, with some improvement of cutaneous lesions using oral tetracycline.18
- Freeze-dried live attenuated measles vaccine administered by injection has been associated with Mucha-Habermann disease.
Most cases of PLEVA cannot be attributed to any one cause and are idiopathic.
No comments:
Post a Comment