Treatment
Medical Care
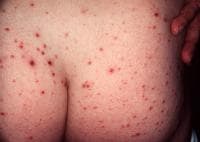
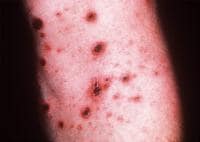
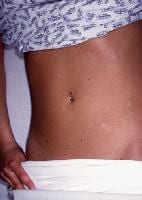
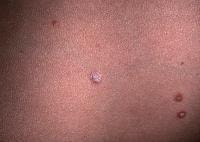
Large ulcerations found in the febrile ulceronecrotic variant of pityriasis lichenoides et varioliformis acuta (PLEVA) require local wound care. Infected lesions may be treated with topical mupirocin and sterile dressing changes twice daily.
No randomized controlled trials of the use of medications have been performed in Mucha-Habermann disease. Since the disease tends towards self-resolution, evaluation of treatments without adequate controls cannot result in useful recommendations. A number of open trials have reported success with light therapy and oral medications.
20Medication
No randomized controlled trials have been performed in Mucha-Habermann disease. Since the disease course tends towards self-resolution, evaluation of treatments without adequate controls cannot result in rational recommendations. Nevertheless, a number of open trials have reported success with light therapy and oral medications.
Phototherapy has been reported useful in the treatment of subacute or chronic disease.
21 Psoralen plus UV-A (PUVA) therapy (150-200 J/cm
2) has been reported, with as many as 4 treatments per week to a total of 30-35 treatments, depending on the patient's skin type. UV-A without psoralens and UV-B may result in clearing. Relapses are not uncommon. Narrow-band ultraviolet B phototherapy and photodynamic therapy have also been reported as effective.
22,23,24,25Case reports suggest the use of multiple oral medications including tetracycline,
18 azithromycin, erythromycin, sulfonamides, dapsone, chloroquine, streptomycin, isoniazid, penicillin, methotrexate (MTX),
26 etretinate, and pentoxifylline. Potent topical corticosteroids may be useful if few lesions are present. Systemic corticosteroids may have a role in severe cases of PLEVA. Despite a lack of randomized controlled trials, oral tetracycline and erythromycin have been prescribed most often in case series.
Antibiotics
May have immunomodulatory activity. Empiric antimicrobial therapy must be comprehensive and should cover all likely pathogens in the context of the clinical setting.
Tetracycline (Sumycin)
Treats gram-positive and gram-negative organisms, as well as mycoplasmal, chlamydial, and rickettsial infections. Inhibits bacterial protein synthesis by binding with 30S and possibly 50S ribosomal subunit(s).
Adult
1-2 g PO divided bid/qid
Pediatric
£ 8 years: Not recommended
>8 years: 25-50 mg/kg PO divided bid/qid
Erythromycin (E.E.S., E-Mycin, Ery-Tab)
Inhibits bacterial growth, possibly by blocking dissociation of peptidyl tRNA from ribosomes causing RNA-dependent protein synthesis to arrest. For treatment of staphylococcal and streptococcal infections.
In children, age, weight, and severity of infection determine proper dosage. When bid dosing is desired, half-total daily dose may be administered q12h. For more severe infections, dose is doubled.
Adult
250 mg PO qid or 333 mg PO q8h
Pediatric
30-50 mg/kg/d PO in divided doses
Psoralens
Tricyclic furocoumarins, when combined with UV radiation, produce DNA photoproducts resulting in suppression of both DNA synthesis and cell division.
Methoxsalen (Oxsoralen-Ultra, 8-MOP)
Inhibits mitosis by binding covalently to pyrimidine bases in DNA when photoactivated by UV-A.
Adult
0.57 mg/kg PO 1.5-2 h before exposure to UV light; treatments should be at least 48 h apart
Pediatric
Not established
Retinoids
Regulate cell growth and proliferation.
Acitretin (Soriatane)
Retinoic acid analog, similar to etretinate and isotretinoin. Etretinate is main metabolite and has demonstrated clinical effects close to those seen with etretinate. Mechanism of action is unknown.
Adult
0.75-1 mg/kg/d PO in divided doses; increase at weekly intervals by 0.25 mg/kg/d up to 1.5 mg/kg/d; establish maintenance dose (0.5-0.75 mg/kg/d) after 8-10 wk of therapy
Pediatric
Not established
Antimetabolites
MTX inhibits dihydrofolate reductase, thereby hindering DNA synthesis and cell reproduction.
Methotrexate (Folex, Rheumatrex)
May suppress immune system. Ameliorates symptoms of inflammation (eg, pain, swelling, stiffness).
Satisfactory response seen in 3-6 wk following administration. Adjust dose gradually to attain satisfactory response.
Adult
0.3 mg/kg/wk PO/IM; not to exceed 20 mg
Pediatric
5-15 mg/m2/wk PO/IM qd or divided tid administered 12 h apart